npj Computational Materials ( IF 9.7 ) Pub Date : 2024-05-08 , DOI: 10.1038/s41524-024-01270-1 Jin-Soo Kim , Juhwan Noh , Jino Im
|
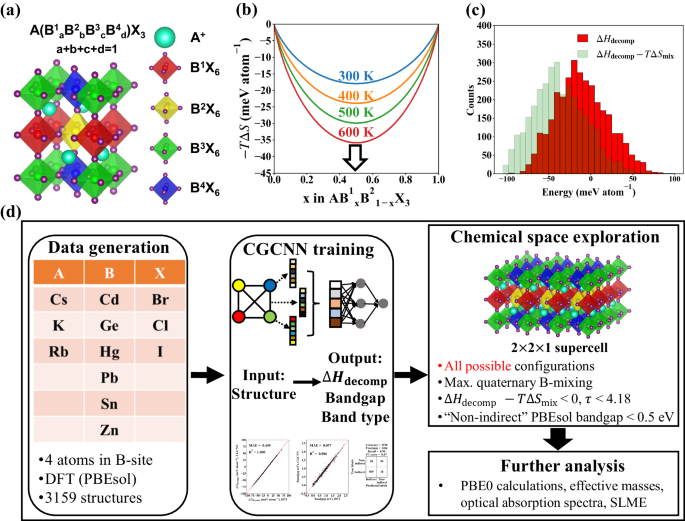
The vast compositional and configurational spaces of multi-element metal halide perovskites (MHPs) result in significant challenges when designing MHPs with promising stability and optoelectronic properties. In this paper, we propose a framework for the design of B-site-alloyed ABX3 MHPs by combining density functional theory (DFT) and machine learning (ML). We performed generalized gradient approximation with Perdew–Burke–Ernzerhof functional for solids (PBEsol) on 3,159 B-site-alloyed perovskite structures using a compositional step of 1/4. Crystal graph convolution neural networks (CGCNNs) were trained on the 3159 DFT datasets to predict the decomposition energy, bandgap, and types of bandgaps. The trained CGCNN models were used to explore the compositional and configurational spaces of 41,400 B-site-alloyed ABX3 MHPs with a compositional step of 1/16, by accessing all possible configurations for each composition. The electronic band structures of the selected compounds were calculated using the hybrid functional (PBE0). Then, we calculated the optical absorption spectra and spectroscopic limited maximum efficiency of the selected compounds. Based on the DFT/ML-combined screening, 10 promising compounds with optimal bandgaps were selected, and from among these 10 compounds, CsGe0.3125Sn0.6875I3 and CsGe0.0625Pb0.3125Sn0.625Br3 were suggested as photon absorbers for single-junction and tandem solar cells, respectively. The design framework presented herein is a good starting point for the design of mixed MHPs for optoelectronic applications.
中文翻译:
基于机器学习的全无机钙钛矿光伏化学空间探索
多元素金属卤化物钙钛矿 (MHP) 的巨大组成和构型空间给设计具有良好稳定性和光电性能的 MHP 带来了重大挑战。在本文中,我们结合密度泛函理论(DFT)和机器学习(ML)提出了一个 B 位合金 ABX 3 MHP的设计框架。我们使用 Perdew-Burke-Ernzerhof 固体泛函 (PBEsol) 对 3,159 个 B 位合金钙钛矿结构进行广义梯度近似,使用 1/4 的组成步骤。晶体图卷积神经网络 (CGCNN) 在 3159 个 DFT 数据集上进行训练,以预测分解能、带隙和带隙类型。经过训练的 CGCNN 模型用于通过访问每种成分的所有可能配置,以 1/16 的成分步骤探索 41,400 个 B 位合金 ABX 3 MHP 的成分和配置空间。使用杂化泛函 (PBE0) 计算所选化合物的电子能带结构。然后,我们计算了所选化合物的光学吸收光谱和光谱极限最大效率。基于DFT/ML组合筛选,选择了10种具有最佳带隙的有前途的化合物,并从这10种化合物中,建议CsGe 0.3125 Sn 0.6875 I 3和CsGe 0.0625 Pb 0.3125 Sn 0.625 Br 3作为单结的光子吸收剂和串联太阳能电池。本文提出的设计框架是光电应用混合 MHP 设计的良好起点。






























 京公网安备 11010802027423号
京公网安备 11010802027423号