Nature Machine Intelligence ( IF 23.8 ) Pub Date : 2024-05-08 , DOI: 10.1038/s42256-024-00837-3 Shuxin Zheng , Jiyan He , Chang Liu , Yu Shi , Ziheng Lu , Weitao Feng , Fusong Ju , Jiaxi Wang , Jianwei Zhu , Yaosen Min , He Zhang , Shidi Tang , Hongxia Hao , Peiran Jin , Chi Chen , Frank Noé , Haiguang Liu , Tie-Yan Liu
|
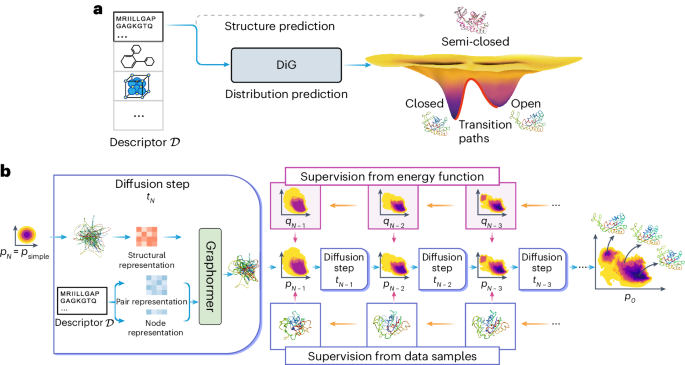
Advances in deep learning have greatly improved structure prediction of molecules. However, many macroscopic observations that are important for real-world applications are not functions of a single molecular structure but rather determined from the equilibrium distribution of structures. Conventional methods for obtaining these distributions, such as molecular dynamics simulation, are computationally expensive and often intractable. Here we introduce a deep learning framework, called Distributional Graphormer (DiG), in an attempt to predict the equilibrium distribution of molecular systems. Inspired by the annealing process in thermodynamics, DiG uses deep neural networks to transform a simple distribution towards the equilibrium distribution, conditioned on a descriptor of a molecular system such as a chemical graph or a protein sequence. This framework enables the efficient generation of diverse conformations and provides estimations of state densities, orders of magnitude faster than conventional methods. We demonstrate applications of DiG on several molecular tasks, including protein conformation sampling, ligand structure sampling, catalyst–adsorbate sampling and property-guided structure generation. DiG presents a substantial advancement in methodology for statistically understanding molecular systems, opening up new research opportunities in the molecular sciences.
中文翻译:
通过深度学习预测分子系统的平衡分布
深度学习的进步极大地改善了分子的结构预测。然而,许多对现实世界应用很重要的宏观观察并不是单个分子结构的函数,而是由结构的平衡分布决定的。获得这些分布的传统方法(例如分子动力学模拟)计算成本昂贵且通常难以处理。在这里,我们引入了一种深度学习框架,称为分布式图形分析器(DiG),试图预测分子系统的平衡分布。受到热力学中退火过程的启发,DiG 使用深度神经网络将简单分布转变为平衡分布,以分子系统描述符(例如化学图或蛋白质序列)为条件。该框架能够有效生成不同的构象,并提供状态密度的估计,比传统方法快几个数量级。我们展示了 DiG 在多项分子任务中的应用,包括蛋白质构象采样、配体结构采样、催化剂吸附物采样和属性引导结构生成。 DiG 在统计学上理解分子系统的方法上取得了重大进展,为分子科学开辟了新的研究机会。






























 京公网安备 11010802027423号
京公网安备 11010802027423号